Vivarium interface basics¶
Overview¶
This notebook introduces the Vivarium interface protocol by working through a simple, qualitative example of transcription/translation, and iterating on model design to add more complexity.
Note: The included examples often skirt best coding practice in favor of simplicity. See Vivarium templates on github for better starting examples: https://github.com/vivarium-collective/vivarium-template
[1]:
#uncomment to install vivarium-core
#!pip install vivarium-core
[2]:
# Imports and Notebook Utilities
import os
import copy
import pylab as plt
import numpy as np
from scipy import constants
import matplotlib.pyplot as plt
import matplotlib
# Process, Deriver, and Composer base classes
from vivarium.core.process import Process, Deriver
from vivarium.core.composer import Composer
from vivarium.core.registry import process_registry
# other vivarium imports
from vivarium.core.engine import Engine, pp
from vivarium.library.units import units
# plotting functions
from vivarium.plots.simulation_output import (
plot_simulation_output, plot_variables)
from vivarium.plots.simulation_output import _save_fig_to_dir as save_fig_to_dir
from vivarium.plots.agents_multigen import plot_agents_multigen
from vivarium.plots.topology import plot_topology
# supress warnings in notebook
import warnings
warnings.filterwarnings('ignore')
AVOGADRO = constants.N_A * 1 / units.mol
# helper functions for composition
def process_in_experiment(
process, settings, initial_state):
composite = process.generate()
return Engine(
composite=composite,
initial_state=initial_state,
**settings)
def composite_in_experiment(
composite, settings, initial_state):
return Engine(
composite=composite,
initial_state=initial_state,
**settings)
store_cmap = matplotlib.cm.get_cmap('Dark2')
dna_color = matplotlib.colors.to_rgba(store_cmap(0))
rna_color = matplotlib.colors.to_rgba(store_cmap(1))
protein_color = matplotlib.colors.to_rgba(store_cmap(2))
global_color = matplotlib.colors.to_rgba(store_cmap(7))
store_colors = {
'DNA': dna_color,
'DNA\n(counts)': dna_color,
'DNA\n(mg/mL)': dna_color,
'mRNA': rna_color,
'mRNA\n(counts)': rna_color,
'mRNA\n(mg/mL)': rna_color,
'Protein': protein_color,
'Protein\n(mg/mL)': protein_color,
'global': global_color}
# plotting configurations
topology_plot_config = {
'settings': {
'coordinates': {
'Tl': (-1,0),
'Tx': (-1,-1),
'Protein': (1,0),
'mRNA': (1,-1),
'DNA': (1,-2),
},
'node_distance': 3.0,
'process_color': 'k',
'store_colors': store_colors,
'dashed_edges': True,
'graph_format': 'vertical',
'color_edges': False},
'out_dir': 'out/'}
plot_var_config = {
'row_height': 2,
'row_padding': 0.2,
'column_width': 10,
'out_dir': 'out'}
Make a Process: minimal transcription¶
Transcription is the biological process by which RNA is synthesized from a DNA template. Here, we define a model with a single mRNA species, \(C\), transcribed from a single gene, \(G\), at transcription rate \(k_{tsc}\). RNA also degrades at rate \(k_{deg}\).
This can written as the difference equation \(\Delta RNA_{C} = (k_{tsc}[Gene_{G}] - k_{deg}[RNA_{C}]) \Delta t\)
Vivarium’s basic elements¶
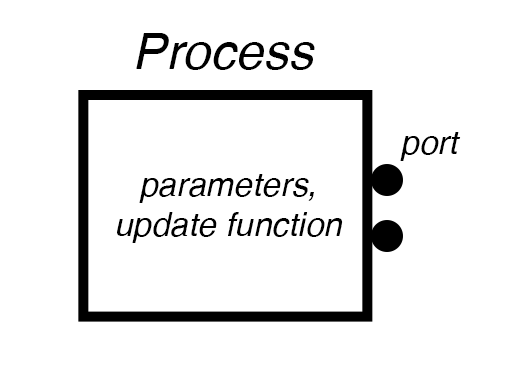
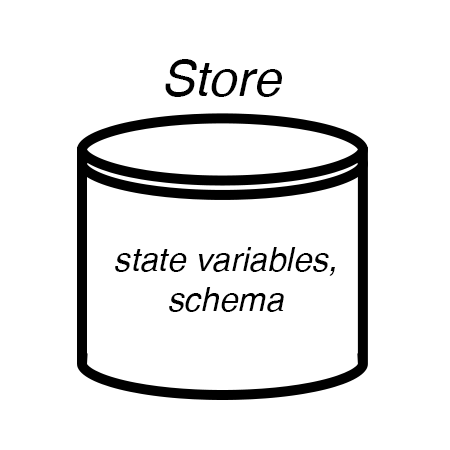
Process interface protocol¶
If standard modeling formats are an “HTML” for systems biology, we need an “interface protocol” such as TCP/IP serves for the internet – a protocol for connecting separate systems into a complex and open-ended network that anyone can contribute to.
Making a dynamical model into a Vivarium Process requires the following protocol: 1. A constructor that accepts parameters and configures the model. 2. A ports_schema that declares the ports and their schema. 3. A next_update that runs the model and returns an update.
Constructor¶
defaultparameters are used in absense of an other provided parameters.The constructor’s
parametersarguments overrides thedefaultparameters.
class Tx(Process):
defaults = {
'ktsc': 1e-2,
'kdeg': 1e-3}
def __init__(self, parameters=None):
super().__init__(parameters)
Ports Schema¶
Ports are the connections by which Process are wired to Stores.
ports_schemadeclares the ports, the variables that go through them, and how those variables operate.Here,
Txdeclares a port formRNAwith variableC, and a port forDNAwith variableG.
def ports_schema(self):
return {
'mRNA': {
'C': {
'_default': 0.0,
'_updater': 'accumulate',
'_divider': 'set',
'_properties': {
'mw': 111.1 units.g / units.mol}},
'DNA': {
'G': {
'_default': 1.0}}
Advanced ports_schema¶
dictionary comprehensions are useful for declaring schema for configured variables.
def ports_schema(self):
molecule_schema = {
'_default': 0.0,
'_emit': True}
return {
'molecules': {
mol_id: molecule_schema
for mol_id in self.parameters['molecules']}}
Advanced ports_schema¶
Use the glob
'*'schema to declare expected sub-store structure, and view all child values of the store:
def ports_schema(self):
schema = {
'port1': {
'*': {
'_default': 1.0
}
}
}
Advanced ports_schema¶
Use the glob
'**'schema to connect to an entire sub-branch, including child nodes, grandchild nodes, etc:
def ports_schema(self):
schema = {
'port1': {
'*': {
'_default': 1.0
}
}
}
Advanced ports_schema¶
Schema methods can also be declared by passing in functions.
The asymmetric_division divider makes molecules in the ‘front’ go to one daughter cell upon division, and those in the ‘back’ go to the other daughter.
def asymmetric_division(value, topology):
if 'front' in topology:
return [value, 0.0]
elif 'back' in topology:
return [0.0, value]
def ports_schema(self):
return {
'front': {
'molecule': {
'_divider': {
'divider': asymmetric_division,
'topology': {'front': ('molecule',)},
}}},
'back': {
'molecule': {
'_divider': {
'divider': asymmetric_division,
'topology': {'back': ('molecule',)},
}}}}
Initial State¶
Each Process MAY provide an
initial_statemethod. This can be retrieved, reconfigured, and passed into a simulation.If left empty, a simulation initializes at the
'_default'values.
def initial_state(self, config):
return {
'DNA': {'G': 1.0},
'mRNA': {'C': 0.0}}
Update Method¶
Retrieve the state variables through the ports.
Run the model for the timestep’s duration.
Return an update to the state variable through the ports.
def next_update(self, states, timestep):
# Retrieve
G = states['DNA']['G']
C = states['mRNA']['C']
# Run
dC = (self.ktsc * G - self.kdeg * C) * timestep
# Return
return {
'mRNA': {
'C': dC}}
Tx: a deterministic transcription process¶
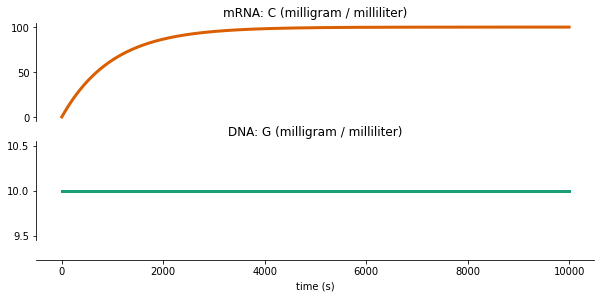
According to BioNumbers, the concentration of DNA in an E. coli cell is on the order of 11-18 mg/mL. The concentration of RNA is 75-120 mg/ml.
[3]:
class Tx(Process):
defaults = {
'ktsc': 1e-2,
'kdeg': 1e-3}
def __init__(self, parameters=None):
super().__init__(parameters)
def ports_schema(self):
return {
'DNA': {
'G': {
'_default': 10 * units.mg / units.mL,
'_updater': 'accumulate',
'_emit': True}},
'mRNA': {
'C': {
'_default': 100 * units.mg / units.mL,
'_updater': 'accumulate',
'_emit': True}}}
def next_update(self, timestep, states):
G = states['DNA']['G']
C = states['mRNA']['C']
dC = (self.parameters['ktsc'] * G - self.parameters['kdeg'] * C) * timestep
return {
'mRNA': {
'C': dC}}
plot Tx topology¶
[4]:
fig = plot_topology(Tx(), filename='tx_topology.pdf', **topology_plot_config)
Writing out/tx_topology.pdf
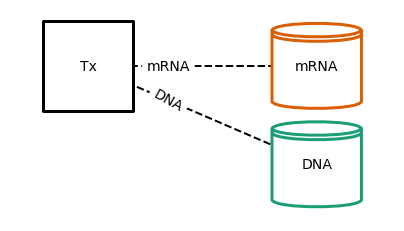
run Tx¶
[5]:
# tsc configuration
tx_config = {'time_step': 10}
tx_sim_settings = {
'experiment_id': 'TX'}
tx_initial_state = {
'mRNA': {'C': 0.0 * units.mg/units.mL}}
tx_plot_config = {
'variables': [
{
'variable': ('mRNA', ('C', 'milligram / milliliter')),
'color': store_colors['mRNA']
},
{
'variable': ('DNA', ('G', 'milligram / milliliter')),
'color': store_colors['DNA']
}],
'filename': 'tx_output.pdf',
**plot_var_config}
[6]:
# initialize
tx_process = Tx(tx_config)
# make the experiment
tx_exp = process_in_experiment(
tx_process, tx_sim_settings, tx_initial_state)
# run
tx_exp.update(10000)
# retrieve the data as a timeseries
tx_output = tx_exp.emitter.get_timeseries()
# plot
fig = plot_variables(tx_output, **tx_plot_config)
Simulation ID: TX
Created: 05/18/2022 at 09:43:24
Completed in 0.283171 seconds
Writing out/tx_output.pdf
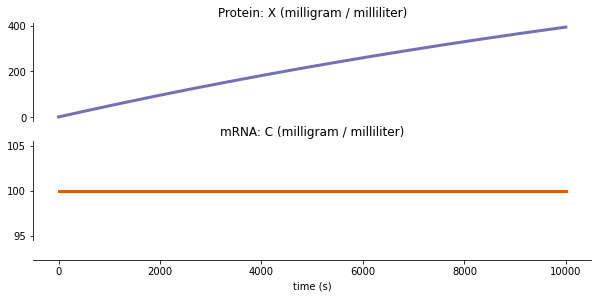
Tl: a deterministic translation process¶
Translation is the biological process by which protein is synthesized with an mRNA template. Here, we define a model with a single protein species, \(Protein_{X}\), transcribed from a single gene, \(RNA_{C}\), at translation rate \(k_{trl}\). Protein also degrades at rate \(k_{deg}\).
This can be represented by a chemical reaction network with the form: * $RNA_{C} \xrightarrow[]{k_{trl}} RNA_{C} + Protein_{X} $ * \(Protein_{X} \xrightarrow[]{k_{deg}} \emptyset\)
According to BioNumbers, the concentration of RNA in an E. coli cell is on the order of 75-120 mg/ml. The concentration of protein is 200-320 mg/ml.
[7]:
class Tl(Process):
defaults = {
'ktrl': 5e-4,
'kdeg': 5e-5}
def ports_schema(self):
return {
'mRNA': {
'C': {
'_default': 100 * units.mg / units.mL,
'_divider': 'split',
'_emit': True}},
'Protein': {
'X': {
'_default': 200 * units.mg / units.mL,
'_divider': 'split',
'_emit': True}}}
def next_update(self, timestep, states):
C = states['mRNA']['C']
X = states['Protein']['X']
dX = (self.parameters['ktrl'] * C - self.parameters['kdeg'] * X) * timestep
return {
'Protein': {
'X': dX}}
run Tl¶
[8]:
# trl configuration
tl_config = {'time_step': 10}
tl_sim_settings = {'experiment_id': 'TL'}
tl_initial_state = {
'Protein': {'X': 0.0 * units.mg / units.mL}}
tl_plot_config = {
'variables': [
{
'variable': ('Protein', ('X', 'milligram / milliliter')),
'color': store_colors['Protein']
},
{
'variable': ('mRNA', ('C', 'milligram / milliliter')),
'color': store_colors['mRNA']
},
],
'filename': 'tl_output.pdf',
**plot_var_config}
[9]:
# initialize
tl_process = Tl(tl_config)
# make the experiment
tl_exp = process_in_experiment(
tl_process, tl_sim_settings, tl_initial_state)
# run
tl_exp.update(10000)
# retrieve the data as a timeseries
tl_output = tl_exp.emitter.get_timeseries()
# plot
fig = plot_variables(tl_output, **tl_plot_config)
Simulation ID: TL
Created: 05/18/2022 at 09:43:26
Completed in 0.297745 seconds
Writing out/tl_output.pdf
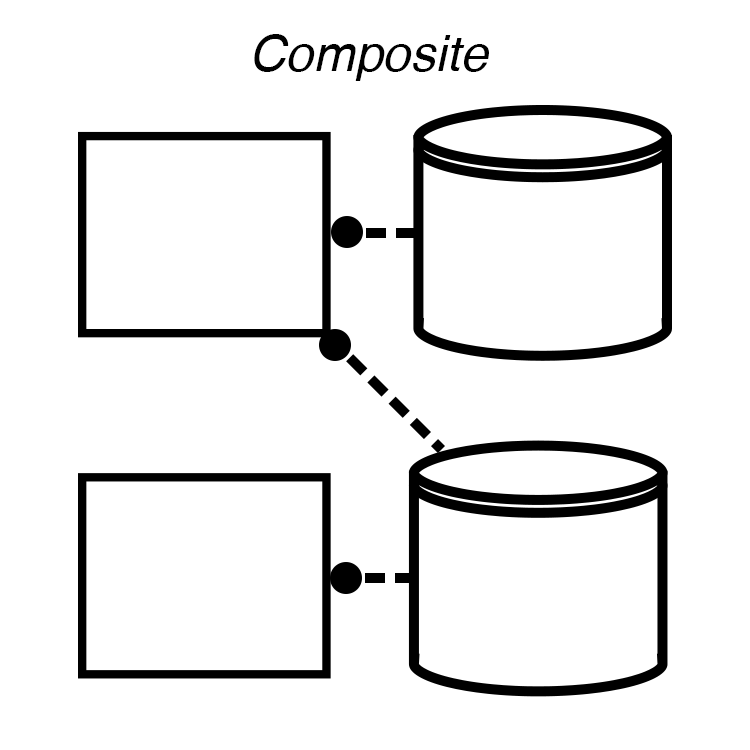
Make a Composite¶
A Composite is a set of Processes and Stores. Vivarium constructs the Stores from the Processes’s port_schema methods and wires them up as instructed by a Topology. The only communication between Processes is through variables in shared Stores.
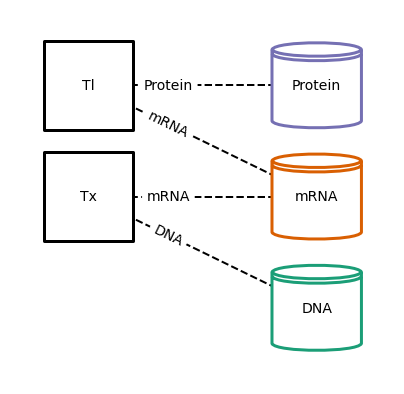
TxTl: a transcription/translation composite¶
We demonstrate composition by combining the Tx and Tl processes.
Composition protocol¶
Composers, which combine processes into composites are implemented with the protocol: 1. A constructor that accepts configuration data, which can override the consituent Processes’ default parameters. 1. A generate_processes method that constructs the Processes, passing model parameters as needed. 2. A generate_topology method that returns the Topology definition which tells Vivarium how to wire up the Processes to Stores.
composite constructor¶
class TxTl(Composer):
defaults = {
'Tx': {
'ktsc': 1e-2},
'Tl': {
'ktrl': 1e-3}}
def __init__(self, config=None):
super().__init__(config)
generate topology¶
Here,
generate_topology()returns the Topology definition that wires these Processes together with 3 Stores, one of them shared.
def generate_topology(self, config):
return {
'Tx': {
'DNA': ('DNA',), # connect TSC's 'DNA' Port to a 'DNA' Store
'mRNA': ('mRNA',)}, # connect TSC's 'mRNA' Port to a 'mRNA' Store
'Tl': {
'mRNA': ('mRNA',), # connect TRL's 'mRNA' Port to the same 'mRNA' Store
'Protein': ('Protein',)}}
advanced generate topology¶
embedding in a hierarchy: to connect to sub-stores in a hierarchy, declare the path through each substore, as done to ‘lipids’.
To connect to supra-stores use
'..'for each level up, as done to'external'.
splitting ports: One port can connect to multiple stores by specifying the path for each variable, as is done to
'transport'.This can be used to re-map variable names, for integration of different models.
def generate_topology(config):
return {
'process_1': {
'lipids': ('organelle', 'membrane', 'lipid'),
'external': ('..', 'environment'),
'transport': {
'glucose_external': ('external', 'glucose'),
'glucose_internal': ('internal', 'glucose'),
}
}}
TxTl Composer¶
[10]:
class TxTl(Composer):
defaults = {
'Tx': {'time_step': 10},
'Tl': {'time_step': 10}}
def generate_processes(self, config):
return {
'Tx': Tx(config['Tx']),
'Tl': Tl(config['Tl'])}
def generate_topology(self, config):
return {
'Tx': {
'DNA': ('DNA',),
'mRNA': ('mRNA',)},
'Tl': {
'mRNA': ('mRNA',),
'Protein': ('Protein',)}}
plot TxTl topology¶
[11]:
txtl_topology_plot_config = copy.deepcopy(topology_plot_config)
txtl_topology_plot_config['settings']['node_distance'] = 2
fig = plot_topology(TxTl(), filename='txtl_topology.pdf', **topology_plot_config)
Writing out/txtl_topology.pdf
run TxTl¶
[12]:
# tsc_trl configuration
txtl_config = {}
txtl_exp_settings = {'experiment_id': 'TXTL'}
txtl_plot_config = {
'variables':[
{
'variable': ('Protein', ('X', 'milligram / milliliter')),
'color': store_colors['Protein']
},
{
'variable': ('mRNA', ('C', 'milligram / milliliter')),
'color': store_colors['mRNA']
},
{
'variable': ('DNA', ('G', 'milligram / milliliter')),
'color': store_colors['DNA']
},
],
'filename': 'txtl_output.pdf',
**plot_var_config}
tl_initial_state = {
'mRNA': {'C': 0.0 * units.mg / units.mL},
'Protein': {'X': 0.0 * units.mg / units.mL}}
[13]:
# construct TxTl
txtl_composite = TxTl(txtl_config).generate()
# make the experiment
txtl_experiment = composite_in_experiment(
txtl_composite, txtl_exp_settings, tl_initial_state)
# run it and retrieve the data that was emitted to the simulation log
txtl_experiment.update(20000)
txtl_output = txtl_experiment.emitter.get_timeseries()
# plot the output
fig = plot_variables(txtl_output, **txtl_plot_config)
Simulation ID: TXTL
Created: 05/18/2022 at 09:43:27
Completed in 0.862072 seconds
Writing out/txtl_output.pdf
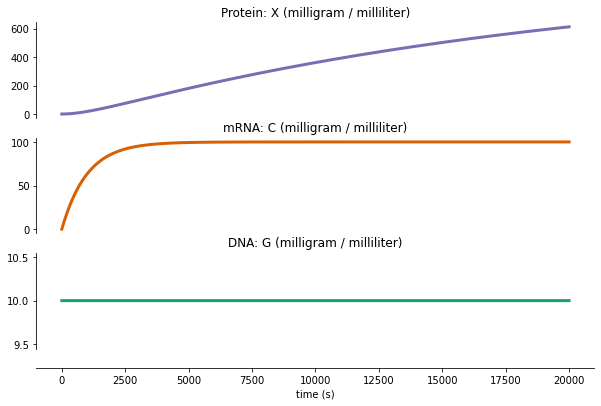
Adding Complexity¶
Process modularity allows modelers to iterate on model design by swapping out different models. We demonstrated this by replacing the deterministic Transcription Process with a Stochastic Transcription Process.
Stochastic transcription requires variable timesteps, which Vivarium accomodates with multi-timestepping.
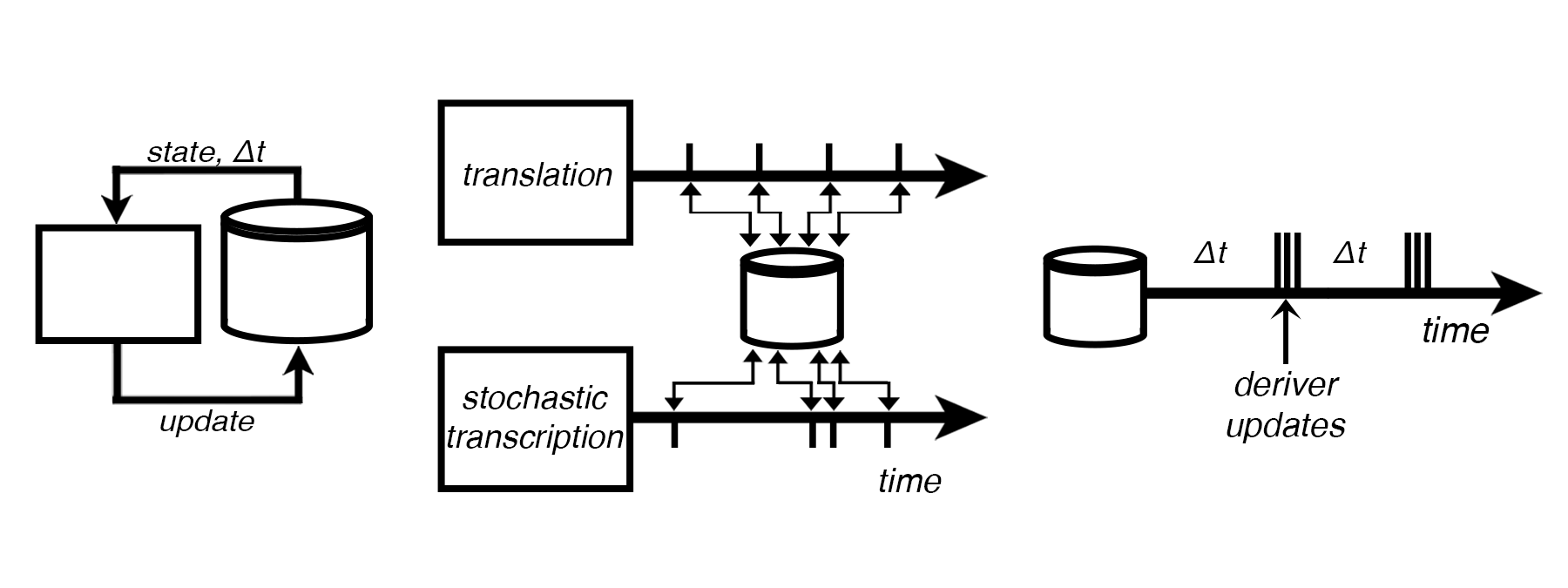
StochasticTx: a stochastic transcription process¶
This process uses the Gillespie algorithm in its next_update() method.
[14]:
stoch_exp_settings = {
'settings': {
'experiment_id': 'stochastic_txtl'},
'initial_state': {
'DNA\n(counts)': {
'G': 1.0
},
'mRNA\n(counts)': {
'C': 0.0
},
'Protein\n(mg/mL)': {
'X': 0.0 * units.mg / units.mL
}}}
stoch_plot_config = {
'variables':[
{
'variable': ('Protein\n(mg/mL)', ('X', 'milligram / milliliter')),
'color': store_colors['Protein'],
'display': 'Protein: X (mg/mL)'},
{
'variable': ('mRNA\n(mg/mL)', ('C', 'milligram / milliliter')),
'color': store_colors['mRNA'],
'display': 'mRNA: C (mg/mL)'},
{
'variable': ('DNA\n(counts)', 'G'),
'color': store_colors['DNA'],
'display': 'DNA: G (counts)'},
],
'filename': 'stochastic_txtl_output.pdf',
**plot_var_config}
[15]:
class StochasticTx(Process):
defaults = {
'ktsc': 1e0,
'kdeg': 1e-3}
def __init__(self, parameters=None):
super().__init__(parameters)
self.ktsc = self.parameters['ktsc']
self.kdeg = self.parameters['kdeg']
self.stoichiometry = np.array([[0, 1], [0, -1]])
self.time_left = None
self.event = None
# initialize the next timestep
initial_state = self.initial_state()
self.calculate_timestep(initial_state)
def initial_state(self, config=None):
return {
'DNA': {
'G': 1.0
},
'mRNA': {
'C': 1.0
}
}
def ports_schema(self):
return {
'DNA': {
'G': {
'_default': 1.0,
'_emit': True}},
'mRNA': {
'C': {
'_default': 1.0,
'_emit': True}}}
def calculate_timestep(self, states):
# retrieve the state values
g = states['DNA']['G']
c = states['mRNA']['C']
array_state = np.array([g, c])
# Calculate propensities
propensities = [
self.ktsc * array_state[0], self.kdeg * array_state[1]]
prop_sum = sum(propensities)
# The wait time is distributed exponentially
self.calculated_timestep = np.random.exponential(scale=prop_sum)
return self.calculated_timestep
def next_reaction(self, x):
"""get the next reaction and return a new state"""
propensities = [self.ktsc * x[0], self.kdeg * x[1]]
prop_sum = sum(propensities)
# Choose the next reaction
r_rxn = np.random.uniform()
i = 0
for i, _ in enumerate(propensities):
if r_rxn < propensities[i] / prop_sum:
# This means propensity i fires
break
x += self.stoichiometry[i]
return x
def next_update(self, timestep, states):
if self.time_left is not None:
if timestep >= self.time_left:
event = self.event
self.event = None
self.time_left = None
return event
self.time_left -= timestep
return {}
# retrieve the state values, put them in array
g = states['DNA']['G']
c = states['mRNA']['C']
array_state = np.array([g, c])
# calculate the next reaction
new_state = self.next_reaction(array_state)
# get delta mRNA
c1 = new_state[1]
d_c = c1 - c
update = {
'mRNA': {
'C': d_c}}
if self.calculated_timestep > timestep:
# didn't get all of our time, store the event for later
self.time_left = self.calculated_timestep - timestep
self.event = update
return {}
# return an update
return {
'mRNA': {
'C': d_c}}
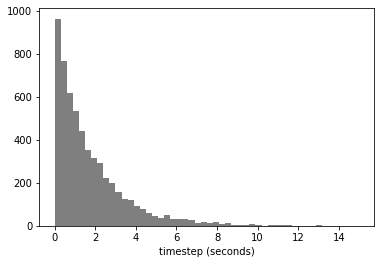
plot variable timesteps¶
[16]:
stoch_tx_process = StochasticTx(tx_config)
stoch_tx_exp = process_in_experiment(stoch_tx_process, **stoch_exp_settings)
stoch_tx_exp.update(10000)
stoch_tx_output = stoch_tx_exp.emitter.get_timeseries()
# calculate the timesteps
times = stoch_tx_output['time']
timesteps = []
for x, y in zip(times[0::], times[1::]):
# if y-x != 1.0:
timesteps.append(y-x)
fig = plt.hist(timesteps, 50, color='tab:gray')
plt.xlabel('timestep (seconds)')
plt.savefig('out/stochastic_timesteps.pdf')
Simulation ID: stochastic_txtl
Created: 05/18/2022 at 09:43:29
Completed in 0.473557 seconds
Auxiliary Processes¶
Connecting different Processes may require addition ‘helper’ Processes to make conversions and adapt their unique requirements different values.
Derivers are a subclass of Process that runs after the other dynamic Processes and derives some states from others.
A concentration deriver convert the counts of the stochastic process to concentrations. This is available in the process_registry
concentrations_deriver = process_registry.access('concentrations_deriver')
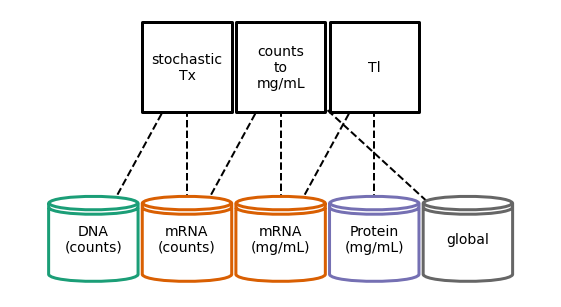
Combining stochastic Tx with deterministic Tl¶
[17]:
# configuration data
mw_config = {'C': 1e8 * units.g / units.mol}
[18]:
class StochasticTxTl(Composer):
defaults = {
'stochastic_Tx': {},
'Tl': {'time_step': 1},
'concs': {
'molecular_weights': mw_config}}
def generate_processes(self, config):
counts_to_concentration = process_registry.access('counts_to_concentration')
return {
'stochastic\nTx': StochasticTx(config['stochastic_Tx']),
'Tl': Tl(config['Tl']),
'counts\nto\nmg/mL': counts_to_concentration(config['concs'])}
def generate_topology(self, config):
return {
'stochastic\nTx': {
'DNA': ('DNA\n(counts)',),
'mRNA': ('mRNA\n(counts)',)
},
'Tl': {
'mRNA': ('mRNA\n(mg/mL)',),
'Protein': ('Protein\n(mg/mL)',)
},
'counts\nto\nmg/mL': {
'global': ('global',),
'input': ('mRNA\n(counts)',),
'output': ('mRNA\n(mg/mL)',)
}}
plot StochasticTxTl topology¶
[19]:
# plot topology after merge
stochastic_topology_plot_config = copy.deepcopy(topology_plot_config)
stochastic_topology_plot_config['settings']['graph_format'] = 'horizontal'
stochastic_topology_plot_config['settings']['coordinates'] = {
'stochastic\nTx': (2, 1),
'counts\nto\nmg/mL': (3, 1),
'Tl': (4, 1),
'DNA\n(counts)': (1,-1),
'mRNA\n(counts)': (2,-1),
'mRNA\n(mg/mL)': (3,-1),
'Protein\n(mg/mL)': (4,-1)}
stochastic_topology_plot_config['settings']['dashed_edges'] = True
stochastic_topology_plot_config['settings']['show_ports'] = False
stochastic_topology_plot_config['settings']['node_distance'] = 2.2
stochastic_txtl = StochasticTxTl()
fig = plot_topology(
stochastic_txtl,
filename='stochastic_txtl_topology.pdf',
**stochastic_topology_plot_config)
Writing out/stochastic_txtl_topology.pdf
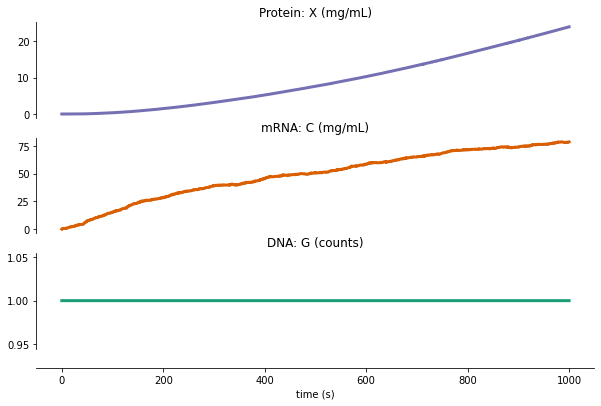
run StochasticTxTl¶
[20]:
# make the experiment
stoch_experiment = composite_in_experiment(stochastic_txtl.generate(), **stoch_exp_settings)
# simulate and retrieve the data
stoch_experiment.update(1000)
stochastic_txtl_output = stoch_experiment.emitter.get_timeseries()
# plot output
fig = plot_variables(stochastic_txtl_output, **stoch_plot_config)
Simulation ID: stochastic_txtl
Created: 05/18/2022 at 09:43:30
Completed in 0.983180 seconds
Writing out/stochastic_txtl_output.pdf
Growth and Division¶
We here extend the Transcription/Translation model with division. This require many instances of the processes to run simultaneously in a single simulation. To support such phenomena, Vivarium adopts an agent-based modeling bigraphical formalism, with embedded compartments that can spawn new compartments during runtime.
Hierarchical Embedding¶
To support this requirement, Processes can be embedded in a hierarchical representation of embedded compartments. Vivarium uses a bigraph formalism – a graph with embeddable nodes that can be placed within other nodes.
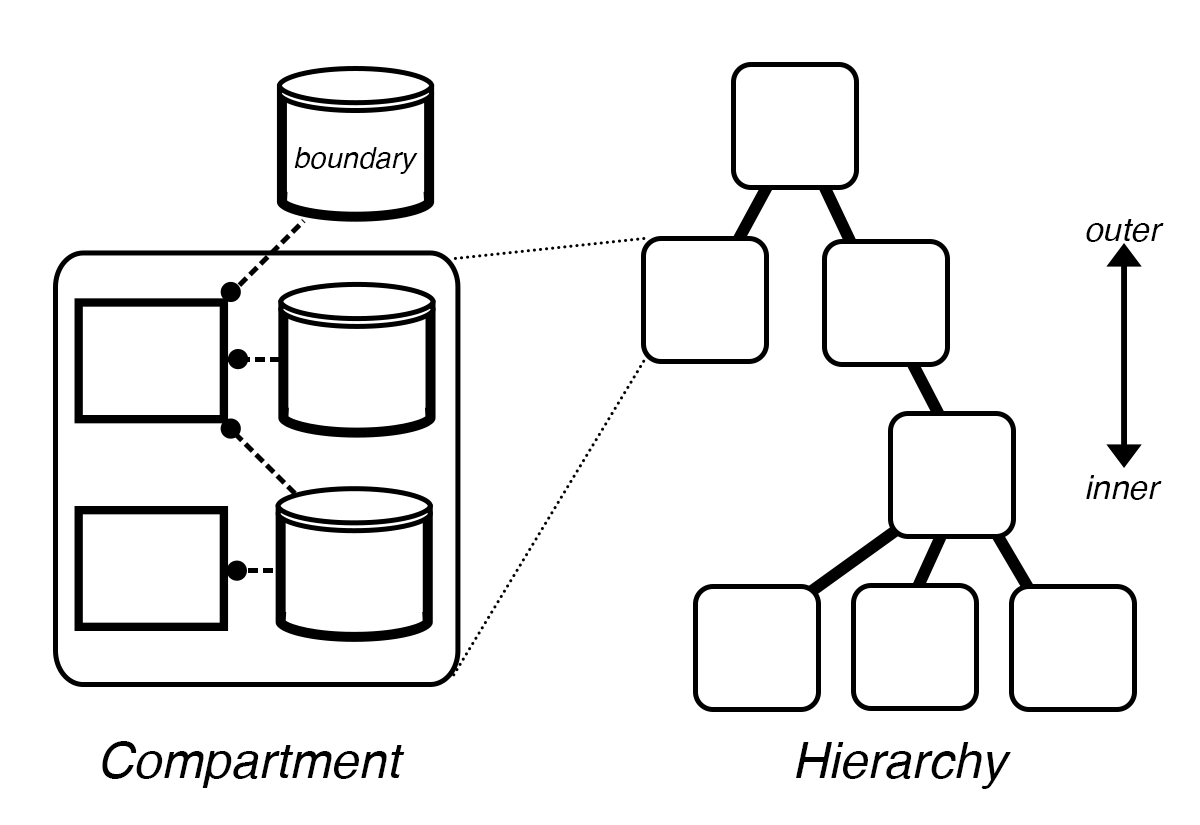
Hierarchy updates¶
The structure of a hierarchy has its own type of constructive dynamics with formation/destruction, merging/division, engulfing/expelling of compartments
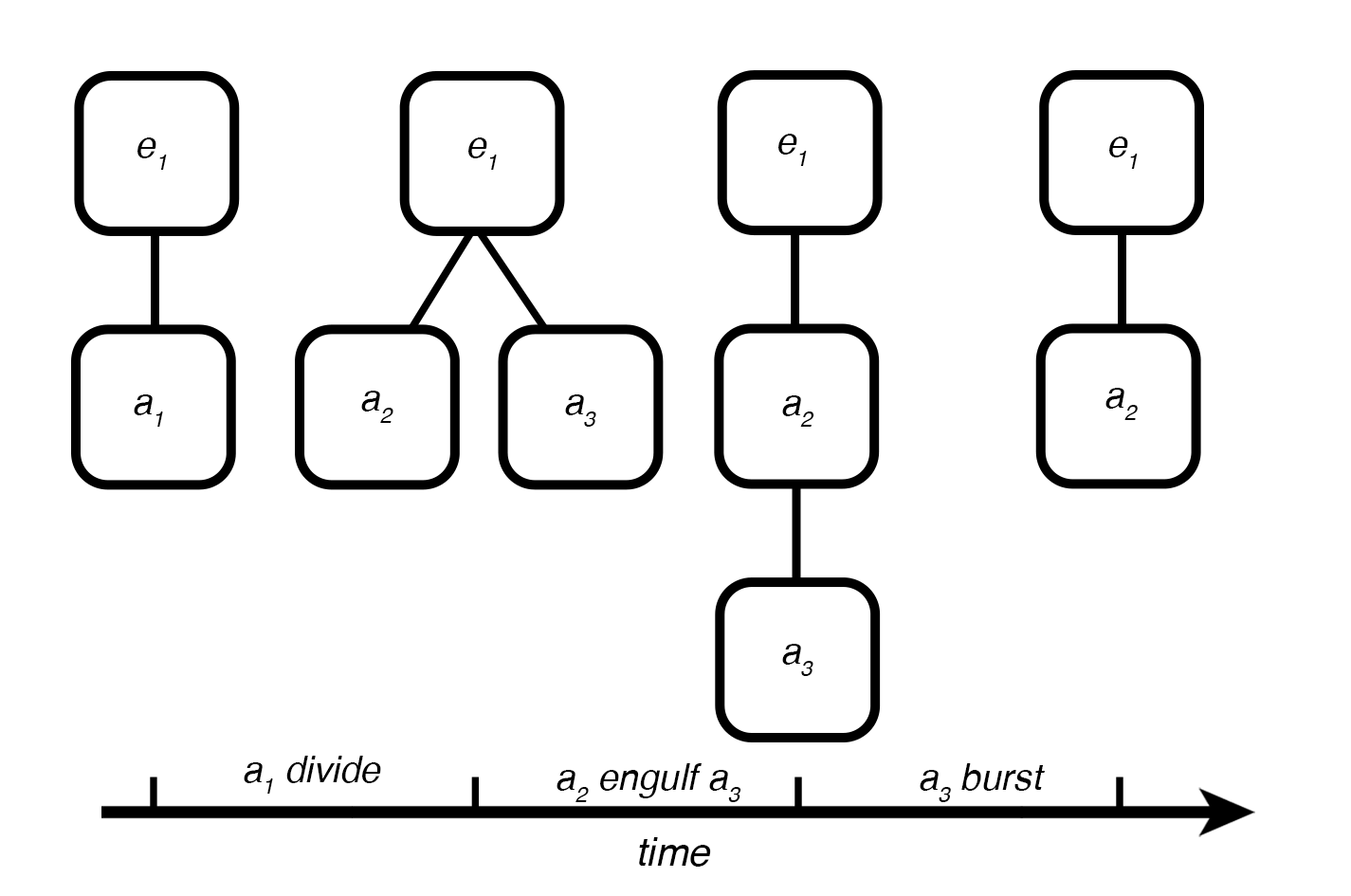
[21]:
# add imported division processes
from vivarium.processes.divide_condition import DivideCondition
from vivarium.processes.meta_division import MetaDivision
from vivarium.processes.growth_rate import GrowthRate
TIMESTEP = 10
class TxTlDivision(Composer):
defaults = {
'stochastic_Tx': {'time_step': TIMESTEP},
'Tl': {'time_step': TIMESTEP},
'concs': {
'molecular_weights': mw_config},
'growth': {
'time_step': 1,
'default_growth_rate': 0.0005,
'default_growth_noise': 0.001,
'variables': ['volume']},
'agent_id': np.random.randint(0, 100),
'divide_condition': {
'threshold': 2.5 * units.fL},
'agents_path': ('..', '..', 'agents',),
'daughter_path': tuple(),
'_schema': {
'concs': {
'input': {'C': {'_divider': 'binomial'}},
'output': {'C': {'_divider': 'set'}},
}
}
}
def generate_processes(self, config):
counts_to_concentration = process_registry.access('counts_to_concentration')
division_config = dict(
daughter_path=config['daughter_path'],
agent_id=config['agent_id'],
composer=self)
return {
'stochastic_Tx': StochasticTx(config['stochastic_Tx']),
'Tl': Tl(config['Tl']),
'concs': counts_to_concentration(config['concs']),
'growth': GrowthRate(config['growth']),
'divide_condition': DivideCondition(config['divide_condition']),
'division': MetaDivision(division_config),
}
def generate_topology(self, config):
return {
'stochastic_Tx': {
'DNA': ('DNA',),
'mRNA': ('RNA_counts',)
},
'Tl': {
'mRNA': ('RNA',),
'Protein': ('Protein',)
},
'concs': {
'global': ('global',),
'input': ('RNA_counts',),
'output': ('RNA',)
},
'growth': {
'variables': ('global',),
'rates': ('rates',)
},
'divide_condition': {
'variable': ('global', 'volume',),
'divide': ('global', 'divide',)},
'division': {
'global': ('global',),
'agents': config['agents_path']}
}
Colony-level processes¶
[22]:
from vivarium.library.units import Quantity
def calculate_volume(value, path, node):
if isinstance(node.value, Quantity) and node.units == units.fL:
return value + node.value
else:
return value
class ColonyVolume(Deriver):
defaults = {
'colony_path': ('..', '..', 'agents')}
def ports_schema(self):
return {
'colony': {
'volume': {
'_default': 1.0 * units.fL,
'_updater': 'set',
'_emit': True}}}
def next_update(self, timestep, states):
return {
'colony': {
'volume': {
'_reduce': {
'reducer': calculate_volume,
'from': self.parameters['colony_path'],
'initial': 0.0 * units.fL}}}}
[23]:
# configure hierarchy
# agent config
agent_id = '0'
agent_config = {'agent_id': agent_id}
# environment config
env_config = {}
# initial state
hierarchy_initial_state = {
'agents': {
agent_id: {
'global': {'volume': 1.2 * units.fL},
'DNA': {'G': 1},
'RNA': {'C': 5 * units.mg / units.mL},
'Protein': {'X': 50 * units.mg / units.mL}}}}
# experiment settings
exp_settings = {
'experiment_id': 'hierarchy_experiment',
'initial_state': hierarchy_initial_state,
'emit_step': 100.0}
# plot config
hierarchy_plot_settings = {
'include_paths': [
('RNA_counts', 'C'),
('RNA', 'C'),
('DNA', 'G'),
('Protein', 'X'),
],
'store_order': ('Protein', 'RNA_counts', 'DNA', 'RNA'),
'titles_map': {
('Protein', 'X',): 'Protein',
('RNA_counts', 'C'): 'RNA',
('DNA', 'G',): 'DNA',
'RNA': 'RNA',
},
'column_width': 6,
'row_height': 1.0,
'stack_column': True,
'tick_label_size': 10,
'linewidth': 1,
'title_size': 10}
colony_plot_config = {
'variables': [('global', ('volume', 'femtoliter'))],
'filename': 'colony_growth.pdf',
**plot_var_config}
# hierarchy topology plot
agent_0_string = 'agents\n0'
agent_1_string = 'agents\n00'
agent_2_string = 'agents\n01'
row_1 = 0
row_2 = -1
row_3 = -2
row_4 = -3
node_space = 0.75
vertical_space=0.9
bump = 0.1
process_column = -0.2
agent_row = -3.2
agent_column = bump/2 #0.5
hierarchy_topology_plot_config = {
'settings': {
'graph_format': 'hierarchy',
'node_size': 6000,
'process_color': 'k',
'store_color': global_color,
'store_colors': {
f'{agent_0_string}\nDNA': dna_color,
f'{agent_0_string}\nRNA': rna_color,
f'{agent_0_string}\nRNA_counts': rna_color,
f'{agent_0_string}\nProtein': protein_color,
},
'dashed_edges': True,
'show_ports': False,
'coordinates': {
# Processes
'ColonyVolume': (2.5, 0),
'agents\n0\nstochastic_Tx': (agent_column, agent_row*vertical_space),
'agents\n0\nTl': (agent_column+node_space, agent_row*vertical_space),
'agents\n0\nconcs': (agent_column+2*node_space, agent_row*vertical_space),
'agents\n0\ndivision': (agent_column+3*node_space, agent_row*vertical_space),
# Stores
'agents': (1.5*node_space, row_1*vertical_space),
'agents\n0': (1.5*node_space, row_2*vertical_space),
'agents\n0\nagents': (1.5*node_space, row_1*vertical_space),
'agents\n0\nDNA': (0, row_3*vertical_space),
'agents\n0\nRNA_counts': (node_space+bump, row_3*vertical_space),
'agents\n0\nRNA': (node_space, (row_3-bump)*vertical_space),
'agents\n0\nProtein': (2*node_space+bump, row_3*vertical_space),
'agents\n0\nglobal': (3*node_space+bump, row_3*vertical_space),
},
'node_labels': {
# Processes
'ColonyVolume': 'Colony\nVolume',
'agents\n0\nstochastic_Tx': 'stochastic\nTx',
'agents\n0\nTl': 'Tl',
'agents\n0\nconcs': 'counts\nto\nmg/mL',
'agents\n0\ngrowth': 'growth',
'agents\n0\ndivision': 'division',
# Stores
# third
'agents\n0': '0',
'agents\n0\nDNA': 'DNA',
'agents\n0\nRNA': 'RNA',
'agents\n0\nrates': 'rates',
'agents\n0\nRNA_counts': '',
'agents\n0\nglobal': 'global',
'agents\n0\nProtein': 'Protein',
# fourth
'agents\n0\nrates\ngrowth_rate': 'growth_rate',
'agents\n0\nrates\ngrowth_noise': 'growth_noise',
},
'remove_nodes': [
'agents\n0\nrates\ngrowth_rate',
'agents\n0\nrates\ngrowth_noise',
'agents\n0\nrates',
'agents\n0\ngrowth',
'agents\n0\ndivide_condition',
'agents\n0\nglobal\ndivide',
'agents\n0\nglobal\nvolume',
]
},
'out_dir': 'out/'
}
# topology plot config for after division
agent_2_dist = 3.5
hierarchy_topology_plot_config2 = copy.deepcopy(hierarchy_topology_plot_config)
# redo coordinates, labels, store_colors, and removal
hierarchy_topology_plot_config2['settings']['node_distance'] = 2.5
hierarchy_topology_plot_config2['settings']['coordinates'] = {}
hierarchy_topology_plot_config2['settings']['node_labels'] = {}
hierarchy_topology_plot_config2['settings']['store_colors'] = {}
# hierarchy_topology_plot_config2['settings']['remove_nodes'] = []
for node_id, coord in hierarchy_topology_plot_config['settings']['coordinates'].items():
if agent_0_string in node_id:
new_id1 = node_id.replace(agent_0_string, agent_1_string)
new_id2 = node_id.replace(agent_0_string, agent_2_string)
hierarchy_topology_plot_config2['settings']['coordinates'][new_id1] = coord
hierarchy_topology_plot_config2['settings']['coordinates'][new_id2] = (coord[0]+agent_2_dist, coord[1])
else:
hierarchy_topology_plot_config2['settings']['coordinates'][node_id] = (coord[0]+agent_2_dist/2, coord[1])
hierarchy_topology_plot_config2['settings']['coordinates']['ColonyVolume'] = (5.5, 0)
for node_id, label in hierarchy_topology_plot_config['settings']['node_labels'].items():
if agent_0_string in node_id:
new_id1 = node_id.replace(agent_0_string, agent_1_string)
new_id2 = node_id.replace(agent_0_string, agent_2_string)
hierarchy_topology_plot_config2['settings']['node_labels'][new_id1] = label
hierarchy_topology_plot_config2['settings']['node_labels'][new_id2] = label
else:
hierarchy_topology_plot_config2['settings']['node_labels'][node_id] = label
hierarchy_topology_plot_config2['settings']['node_labels']['agents\n00'] = '1'
hierarchy_topology_plot_config2['settings']['node_labels']['agents\n01'] = '2'
for node_id, color in hierarchy_topology_plot_config['settings']['store_colors'].items():
if agent_0_string in node_id:
new_id1 = node_id.replace(agent_0_string, agent_1_string)
new_id2 = node_id.replace(agent_0_string, agent_2_string)
hierarchy_topology_plot_config2['settings']['store_colors'][new_id1] = color
hierarchy_topology_plot_config2['settings']['store_colors'][new_id2] = color
else:
hierarchy_topology_plot_config2['settings']['store_colors'][node_id] = color
for node_id in hierarchy_topology_plot_config['settings']['remove_nodes']:
if agent_0_string in node_id:
new_id1 = node_id.replace(agent_0_string, agent_1_string)
new_id2 = node_id.replace(agent_0_string, agent_2_string)
hierarchy_topology_plot_config2['settings']['remove_nodes'].extend([new_id1, new_id2])
use composite.merge to combine colony processes with agents¶
[24]:
# make a txtl composite, embedded under an agents store
txtl_composer = TxTlDivision(agent_config)
hierarchy_composite = txtl_composer.generate(path=('agents', agent_id))
# make a colony composite, and a topology that connects its colony port to agents store
colony_composer = ColonyVolume(env_config)
colony_composite = colony_composer.generate()
colony_topology = {'ColonyVolume': {'colony': ('agents',)}}
# perform merge
hierarchy_composite.merge(composite=colony_composite, topology=colony_topology)
plot hierarchy topology with before division¶
[25]:
fig = plot_topology(
hierarchy_composite,
filename='hierarchy_topology.pdf',
**hierarchy_topology_plot_config)
Writing out/hierarchy_topology.pdf
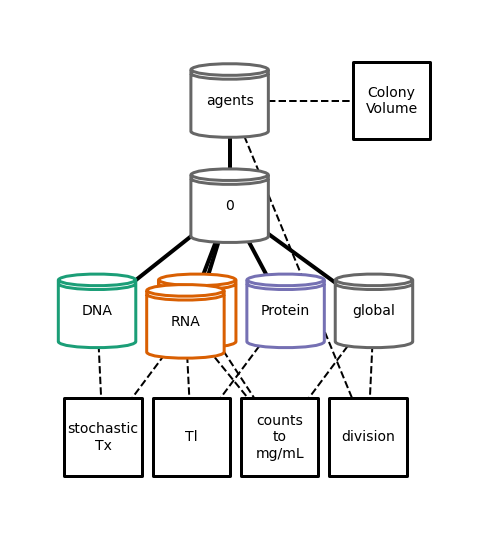
plot hierarchy topology after division¶
[26]:
# initial state
initial_state = {
'agents': {
agent_id: {
'global': {'volume': 1.2 * units.fL},
'DNA': {'G': 1},
'RNA': {'C': 5 * units.mg / units.mL},
'Protein': {'X': 50 * units.mg / units.mL}}}}
# make a copy of the composite
txtl_composite1 = copy.deepcopy(hierarchy_composite)
# make the experiment
hierarchy_experiment1 = composite_in_experiment(
composite=txtl_composite1,
settings={},
initial_state=initial_state)
# run the experiment long enough to divide
hierarchy_experiment1.update(2000)
Simulation ID: a7e6aa2e-d6c9-11ec-8a5b-8c85908ac627
Created: 05/18/2022 at 09:43:33
Completed in 5.25 seconds
[27]:
fig = plot_topology(
txtl_composite1,
filename='hierarchy_topology_2.pdf',
**hierarchy_topology_plot_config2)
Writing out/hierarchy_topology_2.pdf
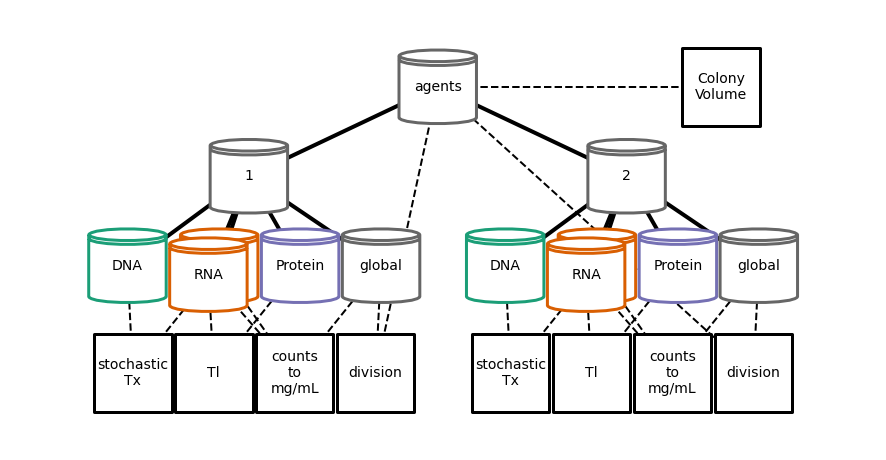
run hierarchy experiment¶
[28]:
# initial state
initial_state = {
'agents': {
agent_id: {
'global': {'volume': 1.2 * units.fL},
'DNA': {'G': 1},
'RNA': {'C': 5 * units.mg / units.mL},
'Protein': {'X': 50 * units.mg / units.mL}}}}
# make the experiment
hierarchy_experiment = composite_in_experiment(
composite=hierarchy_composite,
settings={},
initial_state=initial_state)
# run the experiment
hierarchy_experiment.update(6000)
Simulation ID: ab2e8210-d6c9-11ec-8a5b-8c85908ac627
Created: 05/18/2022 at 09:43:39
Completed in 117.13 seconds
[29]:
# retrieve the data
hierarchy_data = hierarchy_experiment.emitter.get_data_unitless()
path_ts = hierarchy_experiment.emitter.get_path_timeseries()
# add agent colors
dimgray = (0.4,0.4,0.4)
paths = list(path_ts.keys())
agent_ids = set([path[1] for path in paths if '0' in path[1]])
agent_colors = {agent_id: dimgray for agent_id in agent_ids}
hierarchy_plot_settings.update({'agent_colors': agent_colors})
[30]:
# initialize the plot
multigen_fig = plot_agents_multigen(hierarchy_data, hierarchy_plot_settings)
plt.close()
Colony-level metrics¶
[31]:
gd_timeseries = hierarchy_experiment.emitter.get_timeseries()
colony_series = gd_timeseries['agents'][('volume', 'femtoliter')]
time_vec = gd_timeseries['time']
[32]:
# get the RNA_counts axis, to replace with colony volume
allaxes = multigen_fig.get_axes()
ax = None
for axis in allaxes:
if axis.get_title() == 'RNA \nC':
ax = axis
[33]:
# multigen_fig
ax.clear()
ax.plot(time_vec, colony_series, linewidth=1.0, color='darkslategray')
ax.set_xlim([time_vec[0], time_vec[-1]])
ax.set_title('colony volume (fL)', rotation=0)
ax.set_xlabel('time (s)')
ax.spines['bottom'].set_position(('axes', -0.2))
save_fig_to_dir(multigen_fig, 'growth_division_output.pdf')
multigen_fig
Writing out/growth_division_output.pdf
[33]:
